Features on the Synthetic Reads Generator (SyRGen) Dashboard
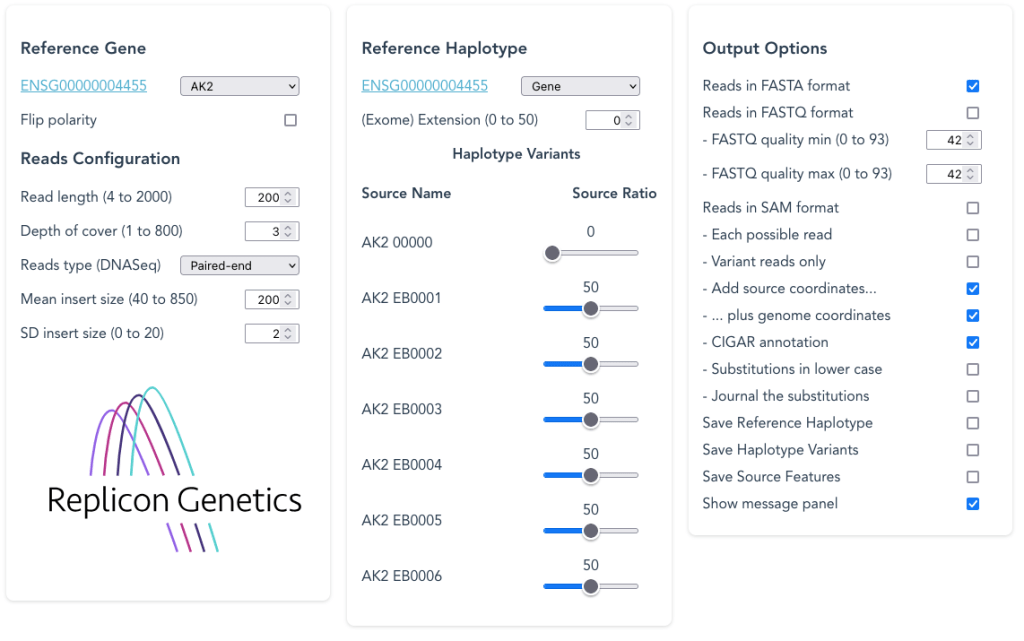
To generate DNA-Seq or RNA-Seq test reads using genomic or exon-based sequence, try the GRCh38 Synthetic Reads Generator
Browser choice
Firefox has been the most reliable browser supporting this application.
The Run button
Before you press the Run button to generate the output files, please note that the Synthetic Reads Generator (SyRGen) generates and saves these files using the capabilities of your browser. It does not download those output files from the internet, but your browser treats them as if they are. You are therefore advised to read the help information about your browser settings for downloading files eg: Firefox ; Chrome ; Microsoft Edge ; Safari, before carrying out your first Run.


On a Mac or PC the output files are typically saved to your “Downloads” folder, unless you specify otherwise. The browser settings may allow you to save to a defined folder, and avoid having to authorise for each file generated.
If the “waiting to complete” button stays in place without any file downloads after a prolonged period of time, please check Troubleshooting notes.
The Headings

These are titles for the dataset and will be different if you have access to a customised or personalised set.

These are hyperlinks to the Replicon Genetics home and information pages on this website.
Selecting Reference Gene

The Reference Gene is the defined gene-region, plus flanking sequence at each end.
The first in the pull-down list here is AK2, with a hyperlink to the Ensembl Gene Summary page for AK2
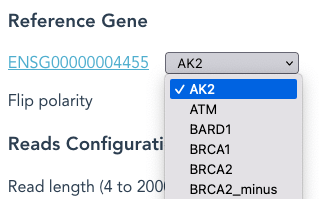
Use the pull-down list to select a different Reference Gene eg: BRCA1.
Each Reference Gene is usually in the forward (+) strand. By selecting “Flip Polarity“, the Reference Haplotype will be defined as the reverse (-) strand.

The same gene-region may appear more than once, as distinct names. eg: BRCA2_minus is the same Locus as BRCA2, but in the reverse (-) orientation.
“Flip Polarity” combined with BRCA2_minus, sets the Reference Haplotype as the forward (+) strand.
Defining the Reference Haplotype
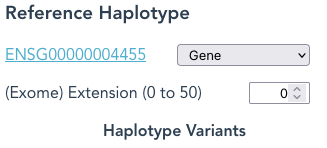
For a sequence to be fragmented into reads, you need to define its Reference Haplotype.
By default the Reference Haplotype is the unspliced Reference Gene; the hyperlink being the same as for the Reference Gene.
The pull-down list offers alternative Reference Haplotypes, the mRNA transcript names, which correspond to identifiers in the Ensembl transcript table.
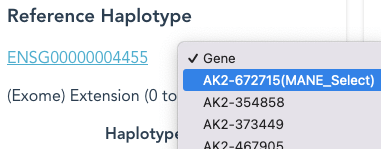
The MANE Select transcript, when defined, is at the top of this list.
With a transcript name selected, the Reference Haplotype will be the (spliced) mRNA transcript; the hyperlink now links to the Ensembl Transcript Summary page, for the selected transcript
By selecting “CDS only” the Reference Haplotype is spliced to the protein-coding sequence only.
If the Reference Haplotype is Gene, the “CDS only” option is hidden.
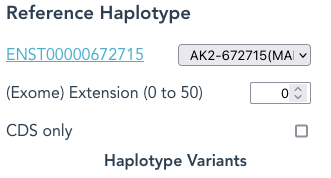
When (Exome) Extension is set higher than the default of 0, eg: 3, then an additional 3 nucleotides of sequence from both 5′ and 3′ ends of each exon will be included as part of each Reference Haplotype, and so appear in reads.
If the Reference Haplotype is Gene, the sequence is trimmed to the defined start and stop of the gene boundary, then increased in length by the value of Extension, at both 5′ and 3′ ends.
Example Ensembl Transcript Summary Page for BRCA1
If the Ensembl summary page includes a button “Show transcript table” instead of “Hide transcript table”, click that button to get the same appearance as above.
The “Name” identifiers are not stable between Ensembl releases, but the Transcript IDs are stable, changing only their version number (after the ‘.’) over time as Ensembl refine their transcript-calling methods.
All the entries in the Ensembl transcript table annotated as “Protein coding” should be present in the Reference Haplotype pull-down list of the application.
The “Non stop decay” and “Nonsense mediated decay” transcripts should also be present.
“Retained intron” or “Processed transcript” entries are not included in the Reference Haplotype pull-down list. Using the source code and data, it is relatively easy to create a version to include these “misc_RNAs”, or any other alternate-splice.
Haplotype Variants: the basics

Each Haplotype Variant may have several Source Names listed eg: “00000”, “hap1”, “hap2”, but other names may be used.
The intention is that the sources each represent a modified Reference Haplotype to indicate definitions of different tissue types, or parental-germline-haplotypes. The Gene_00000 Source Name is a special case to represent the Reference Haplotype which, by definition, means no variants; hence the zeros in the name. Compare BRCA1_00000.gbin to the examples below.
Otherwise each Source Name contains a distinct set of variants: SNV, inserts, deletions. For clarity – take a look at BRCA1_hap1.gbin and BRCA1_hap2.gbin. Selecting Save Source Features as one of the Output Options saves files like this, being the original definitions for the data set.
You cannot change this pre-defined list of Source Names. If you require a different set of Haplotype Variants you may use the “Create a new Haplotype Variant” feature below (see later description)

Alternatively, using the source code and data, you may create a customised set, and each Source Name may be given a distinct suffix name of your choice eg: BRCA1_cust010.
Haplotype Variants Definitions: EGFR demonstration set with exon-19 deletions, including variants not present in public databases

Here, some EGFR Source Names are given prefixes “som1” … “som39” to represent “somatic” sources. Each of these Source Names holds variations, in addition to one of the parental haplotypes, to represent variants that exist in tumour tissue.
Each of the somatic haplotypes in this EGFR set contain a deletion-mutation in exon19, as described in table 2 of “Molecular characteristics and clinical outcomes of EGFR exon 19 indel subtypes to EGFR TKIs in NSCLC patients” by Su et al Oncotargetv.8(67); 2017 Dec 19
Table 2 has been analysed and annotated , with the deletions converted into example Source Names. The allocated number x in somx corresponds to the row number in table2 of the above reference:
Hap1 is parent to: som1; som15; som32
Hap2 is parent to: som14*; som16* ; som39*
The variants with numbers in bold were not present in dbSNP at the date of preparation. Those indicated with an asterisk * were reported in the publication as not detected by available tests.
Defining the relative proportions of haplotypes
To simulate tissue samples, you may adjust the relative Source Ratio between sources, (eg: germline sequence represented by hap1 and hap2) vs (eg: somatic aberrations represented by KRAS G12C). Use the mouse-pointer to drag each slider OR: with the slider selected, use the arrow-buttons on the keyboard.
Examples for KRAS ratio-selections:


By default hap1 and hap2 are set at the same Source Ratio of 50. This means that the number of reads arising from each Source Name, assuming a random-selection, will be approximately the same; 50:50, or 1:1.
By increasing G12C from a default Source Ratio of 50 to 85 (to mimic a higher-than expected ratio from a tumour-tissue sample) the number of G12C reads will now be proportionally higher than for either hap1 or hap2 . In this case the relative Source Ratios of 50:50:85 are re-scaled (normalised) to give actual ratios of 27:27:46 in the output reads.
Should the Source Ratios add up to 100, as with the example of 10:60:30, this equates to percentages more obviously.
- By default the ‘Gene 00000‘ Source Name is set at a Source Ratio of zero and will not generate any reads unless a frequency greater than 0 is set.
- To screen out any individual Source Name, set the Source Ratio to zero.
If you inadvertently set all the Source Ratios to zero, the Run will default to setting a Source Ratio of 1 to the ‘Gene 00000′ Source Name and will proceed to fragment a non-variant Reference Haplotype.
Reads Configuration
The main objective of the output is to provide a file of sequence reads in FASTA format. There are several ways to modify the type of read in that file:

Read length – Changes the length of each sequence read. Enter any value within the range (4 to 2000 in the example).
NB: Extremely low values are useful only for testing purposes and lengthen the time for the program “run” to complete; for paired-end the read length cannot exceed the Mean insert size
Depth of cover – is the number of times each nucleotide should be represented in the FASTA file. SyRGen calculates how many randomly-selected fragments should achieve this value when using the sequence length of the Reference Haplotype (ie: the ‘Gene 00000‘ Haplotype Variant), whether Gene, mRNA or CDS. The application will take a longer time to complete with high Depth-of-Cover values.
Mean insert size – the mean length of the insert in which both reads are located; this is NOT the inner distance between the reads
For the avoidance of doubt: the “Read length” is the length of each read in the pair ; the “fragment length” of a paired-read includes both reads plus the adapters; adapter-sequence is not present in these reads.
SD insert size: the Standard Deviation of the Mean insert size; Combined with the Mean insert size, this is used to draw a random value, using a Gaussian distribution, for each actual insert-length. The method to calculate this uses Numpy.
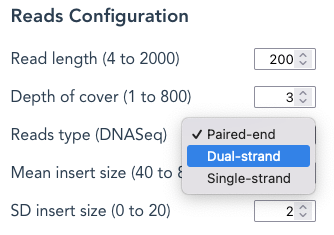
Reads type – A choice between paired-end (default), Dual_strand , or Single-strand.
Paired-end reads
When the Reference Haplotype is Gene, the Haplotype Variant is used as a template, with the first read selected from a random start point that exists in the Haplotype Variant (ie: deleted, substituted or inserted sequence in the variant is not a start-point)
When the Reference Haplotype is an mRNA or CDS, then a read-start-point is selected from all the positions in the mRNA or CDS version of the Haplotype Variant (ie: intron sequence and variants are ignored as a start-point). This is an attempt to model exome sequencing. The start points can be extended into each exome using the (Exome) Extension feature. This is an attempt to model longer capture-baits for the exome-region.
The second read is then calculated from the insert-size. Where either read is close to the end of the Ensembl-defined Gene range, its mate-pair may be outside the Gene range, but will usually be included.
The original Reference Gene, from which each Reference Haplotype is derived, is typically 1000bp longer at each end than the Ensembl-defined Gene range, which should allow inclusion of most mate-pairs.
A mate-pair that would extend beyond the boundary of a Reference Haplotype will exclude both reads in a pair: no single-reads are included in the output, but these unsuccessful ‘unpaired reads’ are reported in the Journal output.
Dual-strand reads : By selecting Dual-strand reads, both forward and reverse-complement (“reverse”) reads are generated, each from a random starting position; except when Each possible read is selected, see below.
Single-strand reads : Only forward reads, each from a random starting point, are represented in the output file; except when Each possible read is selected, see below.

DNASeq vs RNASeq
If the Reference Haplotype is “Gene”: SyRGen will create paired-end genomic-sequence reads, or DNASeq.
If the Reference Haplotype is not “Gene”: SyRGen will create paired-end reads from genomic sequence biased to the coding regions of the Reference Gene, to represent “Exome sequencing”. This is also DNASeq.
If “Paired Ends” is NOT selected and the Reference Haplotype is NOT “Gene”, then the mRNA or CDS sequence is fragmented to represent reads. As these reads exclusively represent RNA sequence, it is labelled as “RNASeq”
Output Options
There are other ways to modify the sequence-reads file, as well as produce other files.
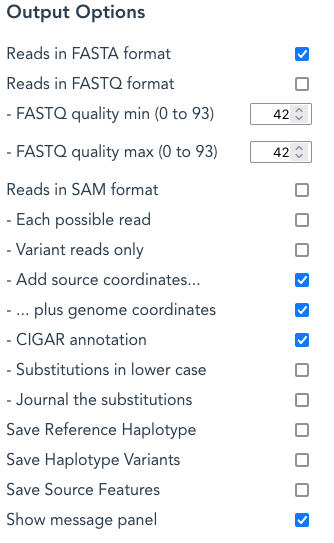
Reads in FASTA format – this option is selected by default, and will generate a file called Gene-ReferenceHaplotype-{mRNA/CDS}_readstype_reads.fasta depending on the Gene, Reference Haplotype, CDS-Only and Reads Type selections
NB: TWO files are produced for paired-end reads
eg: for the ReferenceHaplotype BRCA1 and Gene
BRCA1-gene_paired_DNASeq_reads_R1.fasta and BRCA1-gene_paired_DNASeq_reads_R2.fasta
eg: for the ReferenceHaplotype BRCA1 with transcript 357654
BRCA1-357654_paired_DNASeq_reads_R1.fasta and BRCA1-357654_paired_DNASeq_reads_R2.fasta
For options other than paired ends, the output file names will be one of these, depending on combinations chosen…
or: BRCA1-gene_dual_DNASeq_reads.fasta
or: BRCA1-gene_single_DNASeq_reads.fasta
or: BRCA1-357654-mRNA_dual_RNASeq_reads.fasta;
or: BRCA1-357654-CDS_single_RNASeq_reads.fasta .
By deselecting the Reads in FASTA format tick-box, a reads{}.fasta file will not be produced.
Reads in FASTQ format– select this to create a file named Gene-ReferenceHaplotype-{mRNA/CDS}_readstype_reads.fastq depending on the Gene, Reference Haplotype, CDS-Only and Reads Type selections. NB: TWO files are produced for paired-end reads, as described above for FASTA files
This file contains the same reads in as the FASTA output file(s), but each read includes a FASTQ quality score.
The quality score at each base is a random number between the selected range of the “FASTQ quality min” and “FASTQ quality max”:

To generate real random numbers at each position is very time consuming, especially for high Depth of Cover values. This could lead someone to believe the application had failed, so the default is set with the two values as equal, avoiding the need to generate random numbers.
The application will take a noticeably longer time to complete with FASTQ selected when the two quality values differ in value.
Reads in SAM format – selecting this option creates a SAM-format file (reads aligned against the reference sequence) with the name ReferenceGene-ReferenceHaplotye{-mRNA/CDS}_reads_readtype.sam eg: BRCA1-gene_paired_DNASeq_reads.sam, BRCA1-357654-CDS_RNASeq_reads_dual.sam.
No alignment algorithm has been run, this is just the expected, idealised, result if the alignment were perfectly solved to match all reads to the original reference.
The reference sequence file for SAM output is the one generated by selecting the “Save Reference Haplotype” option with paired-end selected. The file name is ReferenceGene_locseq.fasta eg: BRCA1_locseq.fasta
Or, for RNASeq files, the Reference Haplotype is the spliced mRNA or CDS, and ends with refhap.fasta eg: BRCA1-357654-mRNA_single_refhap.fasta
Use samtools to process the SAM file into BAM files, eg:
% samtools view -@ n -Sb -o BRCA1-gene_paired_reads.bam BRCA1-gene_paired_reads.sam
% samtools sort -O bam -o BRCA1-gene_paired_reads_alignment.bam BRCA1-gene_paired_reads.bam
% samtools index BRCA1-gene_paired_reads_alignment.bam
IGV may reward you with an alignment like this
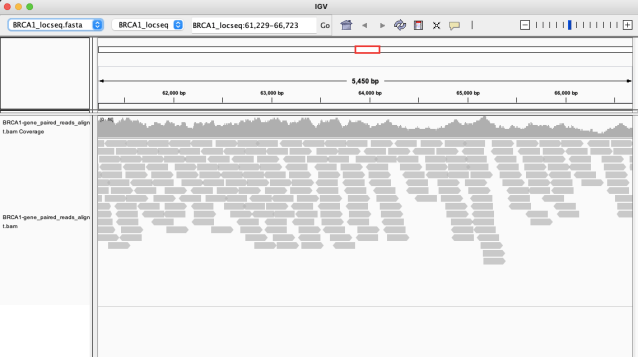
Each possible read –
- When unselected: each read is generated by selecting one Source Name at random from the list, beginning at a random starting point in the sequence of the Haplotype Variant for that Source.
- With Each possible read selected:
- The Depth of cover value is ignored, and each Source Name is taken, in turn, and a read created at each starting point from the first nucleotide position until the (last – Read length) position .
- The length of time it takes to complete a run will increase, because this is similar to selecting a high Depth of cover.
- For Dual-strand reads: both the forward and reverse read are included in the output.
- This option is ignored for Paired-end reads: there are far too many permutations to make this a practicable combination.
Variant reads only – with this option selected, only reads that contain a variant are included in the reads.fasta file.
Add source coordinates/ plus genome coordinates/ CIGAR annotation
Each read in the FASTA file has a header-line given to it, which begins with “>”
The “>” is followed by a unique name for the read “frgn” with an incrementing ordinal number as n. The”frg” part of the name can be changed in a custom set-up.
With all those options turned off the header lines will contain only this unique name eg:
>frg1
>frg2
>frg3
Add source coordinates selected, others unselected, provide header lines like this:
>frg1_hap2 h:115337 r:115337 200M /2
>frg2_hap1 h:65210 r:65210 200M /2
>frg3_hap1 ...
- >the Source Name of the read: hap2 (frg1_hap2)
- the location of the first position in the read as found in:
- h: number; the full-length Haplotype Variant for the Source Name where number is position in the Haplotype Variant sequence once variants are included
- r: number; the full-length Reference Haplotype ie: without variants
- NB: the Reference Sequence is different for Paired-end reads than for the other Reads Types
- the read direction:
- /1 (forward)
- /2 (reverse) is present in Paired Ends and Dual-strand reads
…plus genome coordinates
- annotates the first position in the read, stated as the genomic-mapping location (g:number) on the relevant chromosome.
>frg1_hap2 h:115337 r:115337 g:43158631 200M /2
>frg1_hap2 h:115536 r:115536 g:43158830 200M /1
>frg3_hap1 h:65210 r:65210 g:43108504 200M /2
CIGAR annotation – adds the CIGAR string to the header. With this selected, along with Add source coordinates un-selected, only the CIGAR and read-direction remains in the header:
>frg1 200M/2
>frg1 200M/1
>frg2 200M/2
>frg2 200M/1
More interestingly, a read that includes variants may have a header with a CIGAR like this:
>frg126 74M1X1M1X12M1X1D110M
Substitutions in lower case – when selected, any SNV or inserted nucleotides appear in the read in lower case. Where a delins* definition is used, the inserted part will show as lower case. Deletions are not shown as “..” because this destroys the location-mapping for Add source coordinates.
>frg400 15M8D4I7M1X7M2X5M1X158M CATTGGAACAGAAAGagatTTATCTGtTGTTTGCagTGAAGgAGTACAAAATG
*A delins is simply described as a combination of a deletion and insertion at a given locus, as in the 8D4I CIGAR example below. When explaining this same concept applied to SPDI annotation, NCBI states:
To clarify, using the term “deleted sequence” does not imply that someone is asserting that the mechanism behind the variant was a deletion then an insertion. It only specifies that the same variant sequence would be observed, if this deletion followed by this insertion was applied to the reference sequence.”
https://www.ncbi.nlm.nih.gov/variation/notation/
NB: The delins feature xDyI is the same in both directions, so the reverse-complement CIGAR of
15M8D4I7M1X7M2X5M1X158M is 158M1X5M2X7M1X7M8D4I15M
8D4I is in both, yet the other features are reversed eg: 2X5M1X vs 1X5M2X
NB: The Add source coordinates for xDyI, when it occurs at the start of the read, differs depending on the direction when x > 1!
Journal the substitutions – With this option selected, there are additional journaling records for each variant or gap added. These are retained in the journal file produced for every run (see ‘Other output files produced’, below).
Save Reference Haplotype – Select this to create the following:
When Reads Type is set to Paired-end:
- FASTA format of the unclipped, and un-spliced Reference Gene by the name-format ReferenceGene_locseq.fasta eg: BRCA1_locseq.fasta
- This is the Reference-sequence file for all SAM-format output files for that ReferenceGene for paired-end DNASeq output
- This differs from the Reference Haplotype files for other Reads Type selections, by including more sequence, typically 1000 base pairs at each end, than that defined by the FEATURE locus of the Ensembl-defined Gene-boundary.
When Reads Type is NOT set to Paired-end (ie: Dual-strand or Single-strand) :
- FASTA format of the clipped, and un-spliced Reference Gene by the name-format ReferenceGene-ReferenceHaplotype{-mRNA/CDS}_readtype_REF.fasta eg: BRCA1-gene_single_REF.fasta, BRCA1-357654-mRNA_single_REF.fasta, BRCA1-357654-CDS_dual_REF.fasta
- ‘Clipped’ means: set to the sequence as defined by the FEATURE locus in the Ensembl-defined Gene-boundary. This makes it shorter than the Reference Haplotype created for paired-reads,
- Despite the different prefix-names, each of these REF files holds exactly the same sequence. This has been done to name the reference sequence file in line with the other output file names for the Run, such as the reads themselves..
- This Reference Haplotype is the template prior to
- A) adding variants defined for each Haplotype Variant
- B) splicing
When Reads Type is NOT set to Paired-end, and Reference Haplotype is not Gene, an additional file is produced:
- FASTA format of the clipped and spliced Reference Haplotype called ReferenceGene-ReferenceHaplotype{-mRNA/CDS}_readtype_refhap.fasta eg: BRCA1-357654-mRNA_single_refhap.fasta.
- This ‘refhap’ sequence is the template sequence for each Haplotype Variant before the variants defined in Haplotype Variants are included.
- The word ‘refhap’ is used to distinguish this spliced-template from the reference sequence. Only those variants that lie within this template are included in the reads.
Save Haplotype Variants – select this to create the following files:
When Reads Type is set to Paired-end:
- A FASTA format file that incudes the sequence for each full Haplotype Variant (un-clipped and un-spliced); by the name-format ReferenceGene-gene_readtype_hapvars.fasta eg: BRCA1-gene_paired_hapvars.fasta. These sequences, now including the variants imposed on the original Reference Haplotype, are used as a source during paired-reads-production.
- The sequences differ from those in the hapvars.fasta file produced for other Reads Type selections, by including more sequence, typically 1000 base pairs, at each end, than that defined by the FEATURE locus of the Ensembl-defined Gene-boundary.
- Only the Haplotype Variants with a Source ratio > 0 are included
When Reads Type is NOT set to Paired-end :
- A FASTA format file that incudes the sequence for each full Reference Haplotype (clipped and spliced); by the name-format ReferenceGene-ReferenceHaplotype{-mRNA/CDS}_readtype_hapvars.fasta eg: BRCA1-gene_single_hapvars.fasta, BRCA1-357654-mRNA_single_hapvars.fasta, BRCA1-357654-CDS_single_hapvars.fasta. These sequences, including the variants imposed on the original Reference Haplotype, are used as a source during reads-production.
- Any {X}single_hapvars.fasta and {X}dual_hapvars.fasta will have the same contents, but otherwise different prefixes hold different sequence depending on splicing (none for gene, less sequence for mRNA and even less for cDNA)
- Only the Haplotype Variants with a Source ratio > 0 are included
When CDS only is selected along with an mRNA, (and Reads Type is NOT Paired-end):
- an additional file called Gene-ReferenceHaplotype-CDS_haplotypes_prot.fasta is created eg: BRCA1-357654-CDS_haplotypes_prot.fasta. The file contains the first open-reading frame protein-sequence translation for each sequence in the x_haplotypes.fasta file.
- where the sequences in x_haplotypes.fasta are not on the ‘sense’ strand, the translation is made using the reverse complement of the sequence (based on the polarity and CDS definition in the Reference Source)
- A variant that modifies a sequence which results in the biological destruction of a splice site, is not modelled by this system.
Save Source Features – Select this to create files in Genbank format that contain the Feature Table for the Reference Gene, and each Haplotype Variant Source Names with a Source Ratio > 0.
The list of files created is:
- ReferenceGene_locseq.gbin eg: BRCA1_locseq.gbin, containing the Reference Gene Feature table, eg:lists of mRNA and CDS positions, but no variants and no sequence.
- Each Gene-SourceName.gbin – the definition file for each Haplotype Variant Source Name eg: BRCA1_hap1.gbin , BRCA1_hap2.gbin
- Each ReferenceGene-{gene/mRNA/CDS}_SourceName.gbout with a Source Ratio > 0 eg: BRCA1-gene_hap2.gbout, BRCA1-357654-mRNA_hap2.gbout, contains a modified Feature table from Reference Gene that includes the gaps added to create the Haplotype Variant.
- The additional gap features are annotated in a similar way to variation features originating from public-domain database sources with names like db_xref=”dbSNP:rs12345″
- End-trims are noted as ‘gaps’ by:
- /db_xref=”gap:3-prime downstream trim”
- /db_xref=”gap:5-prime upstream trim”
- excised introns are annotated as ‘gaps’ by their position between exon numbers eg: db_xref=”gap:Intron 22-23″
- the calculated genomic coordinates for each feature, this allows you to validate the input data against external sources, such as dbSNP, Genbank, Ensembl etc:
- /db_xref=”gap:Intron 21-22″
- /global_range=”GRCh37:17:41267797:41276033:1″
- Note: all the features annotated as “intron variants” in BRCA1_hap2.gbin have been removed from the BRCA1 feature table in BRCA1-357654-mRNA_hap2.gbout because they are within an intron region that is now a “gap”. Features present within exon ranges are retained.
Other output files produced –
- Gene-ReferenceHaplotype{mRNA/CDS}_000_Readstype_readme – Gives a summary description of all the files in the output
- eg: BRCA1-gene_paired_DNASeq_000_readme or BRCA1-357654-mRNA_dual_RNASeq_000_readme for mRNA name BRCA1-357654 with “Dual-strand” selected for the Readstype
- Gene-ReferenceHaplotype{-mRNA/CDS}_Readstype_001_journal – Gives a commentary on how the application processed the data,
- eg: BRCA1-gene_paired_DNASeq_001_journal or BRCA1-357654-mRNA_dual_RNASeq_001_journal
- Gene-ReferenceHaplotype{-mRNA/CDS}_001_Readstype_journal.htm – an html version of the above eg: BRCA1–gene_paired_DNASeq_001_journal.htm
- Gene-ReferenceHaplotype{-mRNA/CDS}_002_Readstype_config.txt – This file saves all the dashboard settings.
- eg: BRCA1-gene_paired_DNASeq_002_config.txt or BRCA1-357654-CDS_single_RNASeq_002_config.txt
The readme, journal and config.txt files will be essential to identify and resolve any problems eg: data inconsistencies, program bugs and simple misunderstandings. Please read the contents of these files to see if this resolves any issues before reporting any problems.
Create a New Haplotype Variant

The objective is to select a sub-sequence, the Reference Sequence, which is to be substituted by another, the Variant Sequence. You may select a Reference Sequence at Genomic, Transcript or cDNA level.
The examples presented below show how to replicate the AK2 variants from the HGVS-nomenclature definition , used in Clinvar, given at the cDNA level.
Note that AK2 is coded on the reverse strand, and the method used here is that defining a G > A on the transcript or CDS (anything other than Gene), will actually result in a variant definition on the forward strand as C > T (AK2 EB0003 and dbsnp:rs1192619329)
| Source Name | HGVS expression (CDS-sequence definition) | Comment |
| AK2 EB0001 | NM_001625.4(AK2):c.453del(p.Tyr152fs) implemented in EB0001 as NM_001625.4(AK2):c.452_453CC>C(p.Tyr152fs) | Single-base deletion clinvar:18254 dbSNP:rs1553151177 |
| AK2 EB0002 | NM_001625.4(AK2):c.336_338del(p.Asp113del) | Three-base deletion clinvar:840748 |
| AK2 EB0003 | NM_001625.4(AK2):c.331-1G>A | Splicing-site deletion clinvar:18253 dbSNP:rs1192619329 |
| AK2 EB0004 | NM_001625.4(AK2):c.698_699del(p.Lys233fs) | Deprecated clinvar:1034623 |
| AK2 EB0005 | NM_001625.4(AK2):c.350_402del(p.Lys117Thrfs*32) | Theoretical; 53-base-long deletion |
| AK2 EB0006 | NM_001625.4(AK2):c.406_425+3dup dup=ATCCGAAGAATCACAGGAAGGTA | Theoretical; 23-base duplication crossing a splice-boundary. |
AK2 EB0001: defining c.453del
- Select the MANE_Select Reference Haplotype for AK2, along with CDS Only

- Set 453 on both CDS_Begin and CDS_end.
- The “Absolute” positions are shown as 33014567, which is the GRCh38 mapping for dbSNP: rs1553151177
- The actual Reference Sequence is not currently shown in the Browser version of this application; it is shown in the Python version.
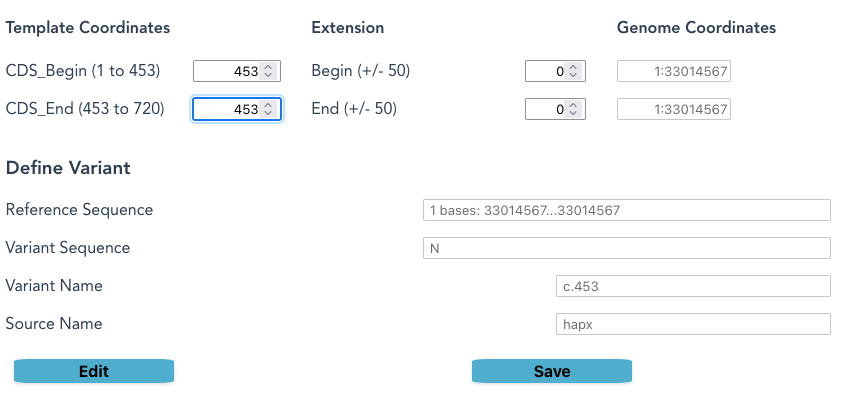
- Click Edit
- In the Variant Sequence box, press Delete, and a “-” character will appear
- Provide a new Source Name and Variant Name, or accept the default
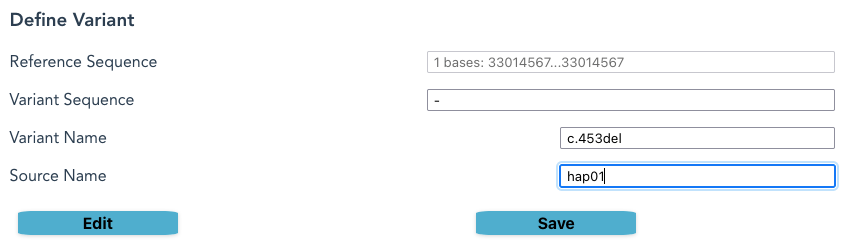
- Click Save.
- If the Variant Sequence differs from the Reference Sequence, a Save will occur.
- If the Source Name differs from the names already in the list, a Save will occur.
- If a Save occurs, the new Haplotype Name appears in the Haplotype Variants column
- If a new name does not appear, go back and check instructions on the previous step: both Variant Sequence and Source Name must be distinct.

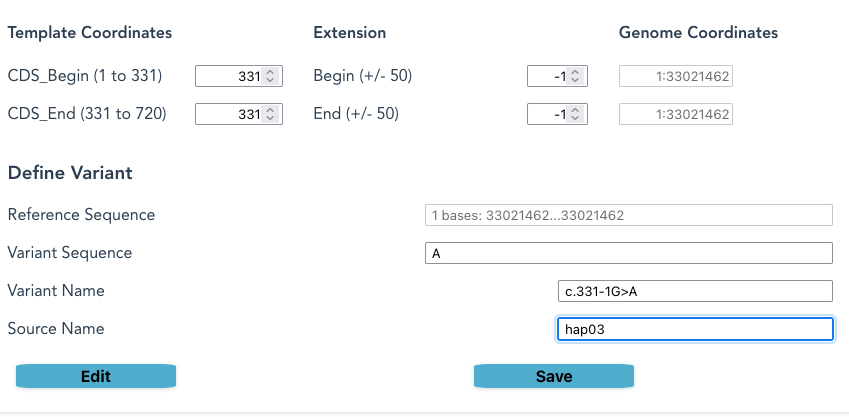
AK2 EB0003: defining c.331-1G>A
Position 331 in the CDS for AK2 is the first base in the third coding region, on a splice-junction.
Nucleotide Sequence (720 nt):
[ATGGCTCCCAGCGTGCCAGCGGCAGAACCCGAGTATCCTAAAGGCATCCGGGCCGTGCTG 60
CTGGGGCCTCCCGGGGCCGGTAAAGGGACCCAG][GCACCCAGATTGGCTGAAAACTTCTGT 120
GTCTGCCATTTAGCTACTGGGGACATGCTGAGGGCCATGGTGGCTTCTGGCTCAGAGCTA 180
GGAAAAAAGCTGAAGGCAACTATGGATGCTGGGAAACTG][GTGAGTGATGAAATGGTAGTG 240
GAGCTCATTGAGAAGAATTTGGAGACCCCCTTGTGCAAAAATGGTTTTCTTCTGGATGGC 300
TTCCCTCGGACTGTGAGGCAGGCAGAAATG][CTCGATGACCTCATGGAGAAGAGGAAAGAG 360
AAGCTTGATTCTGTGATTGAATTCAGCATCCCAGACTCTCTGCTGATCCGAAGAATCACA
GGAAG][GCTGATTCACCCCAAGAGTGGCCGTTCCTACCACGAGGAGTTCAACCCTCCAAAA
GAGCCCATGAAAGATGAC][ATCACCGGGGAACCCTTGATCCGTCGATCAGATGATAATGAA
AAGGCCTTGAAAATCCGCCTGCAAGCCTACCACACTCAAACCACCCCACTCATAGAGTAC
TACAGGAAACGGGGGATCCACTCCGCCATCGATGCATCCCAGACCCCCGATGTCGTGTTC
GCAAGCATCCTAGCAGCCTTCTCCAAAGCCACATGTAAAGACTTGGTTATGTTTATCTAA]
Selecting a number in the Extension field moves the sequence-selection out of the coding region into the adjacent genomic sequence. When on an intron-exon border this means it will select sequence from the intron, as in this example.
For CIITA c.3229_3233+7del
The extension in this example is only at the end of the CDS, creating a very long deletion that includes CDS sequence and 7 bases into an intron, creating a missense in the coding region.
| EB0104 | NM_000246.4:c.3229_3233+7delATGGAGTGAGTG | HGMDPro:unknown |

The Message panel – The contents of the files x_001_y_journal and x_000_y_readme are shown in this panel on the browser as the application runs, and before any files are ready for downloading.
The message panel can be suppressed by de-selecting “show message panel”

Troubleshooting
See release notes for Troubleshooting notes

© Copyright Replicon Genetics & Cary O’Donnell 2021-2025. This and the source code and data are available under the terms of the GNU Affero General Public License version 3 (AGPL-3.0 license
