19th March 2026
An updated web-based version of this application is currently available at GRCh38 Synthetic Reads Generator (SyRGen) but is failing for some browser versions (see troubleshooting section). Firefox working at this date, Chrome is not.
18th February 2026, 13th March 2026:
The browser-based GRCh38 Reads Generator released in October 2025 is failing for some browser versions (see troubleshooting section).
28th October 2025: A web-based version of this application is currently available at GRCh38 Reads Generator
Help page updated 22nd May 2025:
- Sentences using phrases like “contact us for a custom version” changed to referencing the source code and data
- Changes also made to the 18th April previous release notes on this page, which now read “… Reads Generator” instead of “… build; public release”: the same phrase added to the help page.
Edition 31_2: 18th April 2025:
- Two versions available, one for each of the two main genome builds (Firefox recommended)
- GRCh38 Reads Generator – currently unavailable
- GRCh37 Reads Generator – currently unavailable
- Copyright message now includes “… under GNU Affero General Public License version 3”
- Python and JavaScript source available at https://github.com/Replicon-genetics/rg_exploder_shared under the terms of the GNU Affero General Public License version 3 (AGPL-3.0 license)
Edition 31_0: 25th February 2025:
- Two build-versions available, released 25th February 2025
- GRCh38 build; public release (Firefox recommended) – currently unavailable
- GRCh37 build; public release (Firefox recommended) – currently unavailable
- Minor changes to the Genbank-format input files; only noticeable with “Save Source Features” option.
- GRCh37 & GRCh38 versions both have the Source Name for the reference haplotype as locus 000000 (eg: AK2 000000), previously GRCh38 had five zeros instead of six
- GRCh37 : the hyperlink for AK2’s MANE_Select is labelled as GRCh38:ENST00000672715, instead of just ENST00000672715
Edition 30_5: 12th September 2024:
- Two build-versions available, released 12th September 2024
- Paired-end reads for exome (mRNA or CDS) sequence implemented
- FASTQ quality score defaults have been set at 42 for both minimum and maximum value.
- Restored FASTQ quality scores being randomly created for each position.
- When the minimum and maximum values are the same, as in default, the execution speed is faster.
- The output file names have been modified to include “DNASeq” or “RNASeq” depending on the selections made
- “DNASeq” or “RNASeq” is now shown in the “Reads Configuration” section next to “Reads Type”
Edition 20_0: 16th June 2024:
- Two build-versions available, released 16th June 2024
- Paired read start-position: the cutoff has been relaxed; previous version would have reduced the number of reads crossing locus-boundary.
- Depth of Cover maximum value increased from 500 to 800
- FASTQ quality low-score default has been raised from 15 to 35
- FASTQ quality scores are no longer randomly created for each position (see longer description in Help section on how the scores are generated).
- This is because of the previous very long run time for high Depth-of-Cover, which makes it look as though the application is not working.
- Next release objective: paired-end reads for exome (mRNA or CDS) sequence.
Edition 27_05: 23rd May 2024 :
- Two build-versions available released 23rd May 2024
- When “Variant Reads only” is selected with Paired-end:
- Where one of the paired reads is variant and the other is not, both reads are saved, not just the variant one.
- SAM format PNEXT value correction
- Some changes to the text in the Journal and readme files, intended to improve clarity
Edition 27_03: 27th April 2024 :
- Two build-versions available released 27th April 2024
- Corrections to paired-end sequence-generation
- SAM format corrections: PNEXT, FLAG, @SQ line; SAM now 1-based mapping
- PNEXT is, unintentionally, +1 higher than it should be (fix next release) 21-May
- Change in Edition numbering from incrementing the release number to actual software version
Edition 6_1: 6th March 2024 :
- New feature “Create a New Haplotype Variant” allows you to define a new variant, and then include that in the list of Haplotype Variants to be fragmented into reads.
- New genes and variants: AK2, CIITA, NF1 courtesy of Eleanor Baker at North West Genomic Laboratory Hub
- Changes in naming and terminology as listed below
Changes in naming and terminology:
- Mapping “positions” are now called “coordinates”
- “Source coordinates” replaces “Source positions”
- “Genome coordinates” replaces “Absolute position”
- “Reference Haplotype” replaces “Haplotype Definition” is now
- “Reference Gene” replaces “Reference Source”
- “Gene” replaces “Locus” when referring to the defined Ensembl gene-region
- The coordinates included in the FASTA headers are now presented as
- h:(haplotype) r:(reference) g:(genomic) instead of:
- h:(haplotype) r:(reference) a:(absolute)
- eg: >frg1_EB0101 h:15598 r:15598 g:10985652 200M /1
See Troubleshooting for other issues
Edition 0005_02 22 Sep 2022:
There was an error in the two KRAS data sets defining polarity.
Fixes were made to protein translation on haplotypes to ensure the polarity/sense combination is correctly taken into account.
Help pages all updated to current features and output file names.
See Troubleshooting section below
Edition 5_1 23rd August 2022:
Multiple additional features including: Paired-ends sequencing; all loci are now in the forward (+) strand, previously the polarity corresponded with the sense strand of the CDS for that locus; corrections to CIGAR features and mapping positions; terminology changes.
Edition 5 25th September 2021: Transcripts in the Transcript pull-down list are labeled with NMD (nonsense-mediated decay) and NSD (non-stop decay) if known at the time of preparation. (Not available in later releases)
Bug fix to the output files locus_variant.gbout : the db_xref features were not being saved in the correct Genbank style for variants.
Bug-fix to overlap-trimming of variants: some del-ins variant definitions that overlapped intron-exon boundaries were incorrectly masked.
Edition 4 ; Release date: 26th July 2021
When “Save Variant Sequences” is selected:
- The nucleotide file has been changed from x_mut.fasta to x_var.fasta
- There is a new output file x_var.prot, which is a protein translation of x_var.fasta. This is only produced when “CDS” is selected AND “Exome Extension” is set to zero
- The help pages have been largely updated, but example output files, notably their new names, are incomplete
Edition 3; Release date: 21st July 2021
What has changed from edition2 – example links below are for ATM
- “Selected Reference” is now “Locus” and it hyperlinks to the “LRG” entry
- “Reference Sequence” is now “Transcript” and hyperlinks to Ensembl
- If “Genomic”, the hyperlink shows the “gene” page
- If a transcript, it shows the “transcript” page.
- Try looking at the hyperlinked Ensembl pages with “Show transcript table” selected: you should see the “Mane Select” ‘flag’ on the right-hand column
- The “Transcript” selection has the “Mane Select” transcript as the first item in the list.
- The Mane Select transcript is taken from the Ensembl ‘flag’ – many of these are currently missing from LRG.
- If the transcript is listed in LRG as “t1”, “t2” etc, the t-number is included in the name of the transcript
- If you “Run” with a named transcript the introns are removed
- If you “Run” with the transcript set to “Genomic” you get the Ensembl defined gene-range (it previously included 250bp or 500bp beyond this, depending on the dataset) including introns.
- There is a “CDS only” option that clips UTRs from the transcript
- The “Exome Extension” allows up to 50 instead of 9: it adds the selected number of nucleotides at both 5′ and 3′ ends of each splice-junction (this includes the CDS “junctions”), or gene-range.
- Using the Ensembl “Name” for transcripts such as “ATM-201” has been abandoned as these are not stable. Instead a clipped version of the Ensembl Transcript ID is used. It is easy to use any other identifier in its place eg; NM_identifiers
- The help pages have not been updated for this version
Troubleshooting
1. A page hang

This is when the Run button remains in the wait state without any file downloads for a prolonged period of time.
First: if you are using any other browser than Firefox, please try using Firefox instead. See end of this section for more detail.
Second: this may be due to a combination of options that are known to increase the processing time, as documented on the Help page eg: low Read length values; high Depth of Cover values; FASTQ and SAM output. The generation of very large files may take several minutes.
Test this by refreshing the browser and Run again, but using the default settings.
Results should appear within a minute with the defaults, so return to your options and consider changing the values in steps, exploring the relationship between the values and time taken.
Third: if you are familiar with looking at the JS console, you might see some errors there. If so, please report this when contacting a developer.
2. Reads, or other files, not generated
First: check that your expected options, such as Reads in FASTA format is selected and not deselected unintentionally:


Another example is where the Source Ratio of Haplotypes has been set to zero:
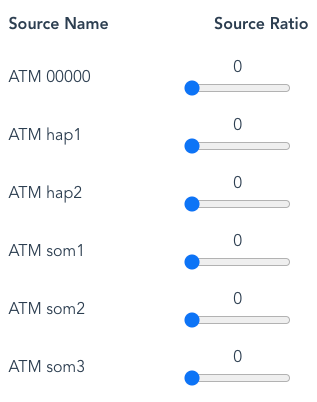
By default this will generate sequence reads from ATM_00000, the reference haplotype, so all reads will lack variants. If “Variant Reads only” has been selected, there will be no output.
Second: If you have selected Paired-end reads, then selections in Reference Haplotype other than Gene will be ignored; CDS only is also ignored in this case.
The following selections of Reference Haplotype are equivalent with Paired-end selected.

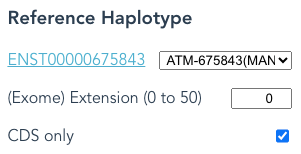
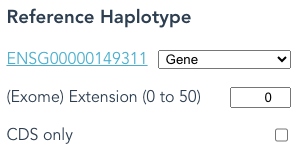
Switch Reads Type to something else to see the difference in output files.
Third: read information in the Message Panel, just below the Copyright message.
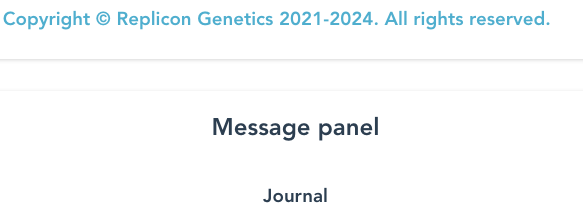
This has two sections which you may need to scroll down to read:
- “Journal”: a verbose account of the processing steps
- “Readme”: a list of all the input and output file
The application will stop if it encounters problems like missing data files and incompatible data sets (this would be unintentional!) and report this eg:
Reading 'Reference Haplotype' file ATM_hap1.gb
“Incompatible GRCh build, chromosome or polarity for sequence: GRCh37:11:1 vs variants: GRCh38:11:1”
“Unable to read a minimum of 1 Reference Haplotype”
In this case select “Save Source Features” and see if the content of the input files is really incompatible eg: compare the files ATM_locseq.gb and ATM_hap1.gbin, by looking at the ACCESSION line:
ATM_locseq.gbin: ACCESSION chromosome:GRCh38:11:108222044:108370102:1 ATM_hap1.gbin: ACCESSION chromosome:GRCh38:11:108222332:108369602:1
If the GRCh versions do not agree, or the genome ranges mismatch, or anything else looks wrong in the data set, please contact a developer.
Browsers
Browser issues February 2026:
As described below: without regular refreshing of the npm build to incorporate browser updates, the provided browser-version will cease to perform correctly. This has already happened for Chrome.
Previous browser issues identified 08-Nov-2022;
Chrome version 99.0.4844.83 (Official Build) (x86_64), (22-Mar 2022 download), AND Version 105.0.5195.110 (Official Build) (x86_64) (08-Nov-2022 download) AND Version 109.0.5414.119 (January 2023) produce the following warning messages in the console:
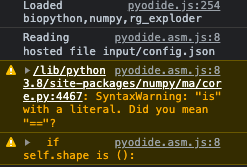
but the application works correctly.
Chrome Version 107.0.5304.110 (Official Build) (x86_64) , (08-Nov-2022 download) produces this error, and the application fails:
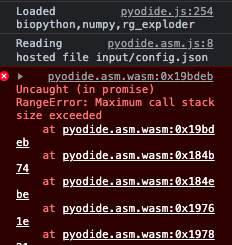
Firefox versions available in November 2022 failed similarly, but Firefox 109.0 (Jan 2023) worked
Actual & proposed solutions:
- When preparing a new AWS release, update the browser list for npm using:
- npx browserslist@latest –update-db
- Status: February, April & October 2025 updated
- Update all generating applications to the latest versions (Python, Biopython, Pyodide, Docker, npm). Status: not implemented
© Copyright Replicon Genetics & Cary O’Donnell 2021-2026 & available under the terms of the GNU Affero General Public License version 3 (AGPL-3.0 license)
